How to design a regulatory strategy to optimize registration of Advanced Therapy Medicinal Products (ATMPs) with European Medicines Agency ?

22 March 2022
BLUEREG GROUP
Advanced Therapy Medicinal Products (ATMP) are promises medicines for untreatable and high burden diseases where conventional approaches are inadequate such as cancer, cardiovascular diseases, musculoskeletal conditions, or immune system disorders1. To allow patients timely access to these new therapies a regulatory environment encouraging innovation and safeguarding public health was needed. The European Union (EU) regulation (EC) No 1394/2007 came into force in 2008, providing a common regulatory framework for ATMPs. The main benefits of this regulation are the mandatory use of the centralized procedure to market an ATMP in Europe and the creation of the Committee for Advanced Therapies (CAT). This European Medicines Agency (EMA) committee brings together expertise of experts from the 27 EU member states responsible for assessing quality, safety, and efficacy of ATMPs.
Development of ATMPs is complex too due to their nature. A tailored and risk-based approach is therefore required throughout the development to determine the extent of quality, non-clinical and clinical data to be included in the marketing authorization application (MAA). Although optional, this approach is strongly recommended to build the marketing authorization application dossier. Early discussions with the CAT are also key for a well-designed regulatory strategy to overcome all the challenges an applicant can face to market an ATMP. Regulatory tools have been developed by the EMA to help applicants designing the most appropriate regulatory strategy for their products and promote early discussions with the EMA to get a successful MAA.
Most ATMPs are developed by micro, small and medium companies as well as universities and academia. Their regulatory experience and knowledge of the EU regulation are often limited. An experienced third party is highly recommended to navigate through the regulation and define the most-appropriate regulatory strategy.
This article outlines the EMA regulatory tools available to successfully market your ATMP in Europe and the high interest of using an experienced third party.
I Regulatory tools for early interactions with the EMA

Interactions with the EMA can start at a very early stage of the product development, generally at the non-clinical stage. They are crucial to define the best strategy. These interactions can be CAT specific tasks (such as ATMP classification and ATMP certification) and CAT non-specific tasks (such as innovation task force briefing meeting and scientific advice). They are optional and some of them are free of charge.
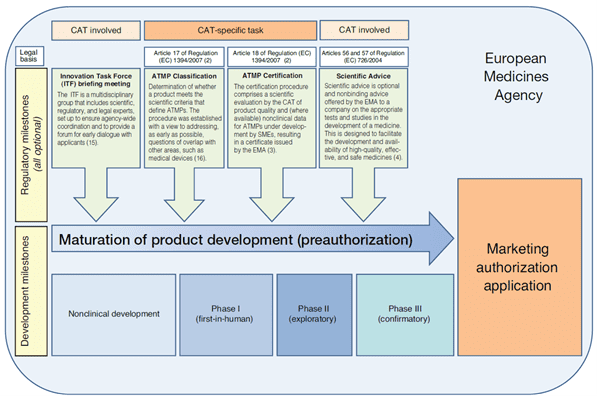
Figure 1: Early interactions with the CAT2
1. Innovation Task Force Briefing Meeting
Innovation Task Force (ITF) is an EMA multidisciplinary group that includes scientific, regulatory, and legal experts from the CAT and other committees. ITF Briefing Meetings provide a forum for early informal exchange of information with regulators generally at the non-clinical stage to cover regulatory, technical, and scientific issues. Guidance, development process as well as complementation and reinforcement of existing formal procedures are also discussed.
These meetings are mainly for micro, small and medium sized companies as well as academics and researchers. They are free of charge.
2. ATMP classification
The ATMP classification is a scientific recommendation led by the CAT to determine whether the assessed product fulfills one of the ATMP definitions as laid out in Regulation (EC) No 1394/2007 and part IV of annex I to directive 2001/83/EC.
There are three types of ATMPs:
- Gene therapy medicinal products: these products contain genes that lead to a therapeutic, prophylactic, or diagnostic effect. They work by inserting 'recombinant' genes into the body, usually to treat a variety of diseases, including genetic disorders, cancer, or long-term diseases. A recombinant gene is a stretch of DNA that is created in the laboratory, bringing together DNA from different sources.
- Somatic cell therapy medicinal products: these products contain cells or tissues that have been manipulated to change their biological characteristics or cells or tissues not intended to be used for the same essential functions in the body. They can be used to cure, diagnose, or prevent diseases.
- Tissue engineered medicinal products: these products consist of, or contain living cells, tissues or organs intended for implantation in the human body in order to replace an impaired function of tissue or organ or for maintaining such function in case it is present already.
Some ATMPs are referred to as “combined ATMPs”: they contain one or more medical devices as an integral part of the medicine such as cells embedded in a biodegradable matrix or scaffold.
The criteria for ATMPs classification are set out in Article 17 of Regulation (EC) No 1394/2007. The ATMP classification is assessed by the CAT on request of and on basis of information provided by a developer of a product. Upon receipt of a valid request, the CAT delivers a scientific recommendation on an ATMP classification after consultation with the European Commission within 60 days. The CAT recommendation is not binding.
This procedure is voluntary and free of charge. The European legislation provides for scientific and financial incentives for developers of ATMPs.
The ATMP classification is an opportunity to initiate an early and tailored dialogue on product development with regulators and provide clarity on the development path and regulatory guidance to be followed.
Non-confidential summaries of previous ATMP classifications are available on the EMA website.
3. ATMP certification
The ATMP certification procedure mentioned in Article 18 of Regulation (EC) No 1394/2007 is a scientific evaluation led by the CAT of quality data and, when available, non-clinical data that have been generated at any stage of the ATMP development process and check that they are on the right track for a successful development. The evaluation and certification procedure takes 90 days.
The ATMP certification procedure is also an opportunity to identify any potential issues that can be addressed before submitting a MAA.
This procedure is applicable to ATMPs under development for micro, small and medium sized enterprises only.
4. PRIORITY MEDICINES
The PRIORITY MEDICINES scheme, also known as PRIME, is a scheme launched in March 2016 by the EMA to enhance support for the development of medicines that target an unmet medical need with a high public health potential.
This scheme is voluntary and allows enhanced and early interactions between developers of medicines and the EMA to optimize development plans to generate robust data on the medicine’s benefits and risks. The PRIME designation allows accelerated assessment of medicine applications.
The PRIME designation is granted when the two following criteria are met:
- The medicine targets an unmet medical need.
- A justification must be provided to explain what the unmet need is and how it will be addressed with evidence.
Once the PRIME designation has been granted to a candidate medicine, there are several benefits for the sponsor during development:
- A rapporteur from the CAT is appointed by the EMA to provide continuous support and guidance that would allow the sponsor to build a robust data package for a MAA
- The EMA organizes a kick-off meeting with the CAT rapporteur and a multidisciplinary group of experts to provide preliminary guidance on development plan, discuss key development steps and regulatory strategy.
- The EMA assigns a dedicated single contact point who will coordinate regulatory support throughout the scheme
- The EMA provides scientific advice at key development milestones and on key issues.
- Confirmation of accelerated assessment at the time of MAA
The FDA has a similar designation to expedite development and review of drugs that are intended to treat serious conditions and preliminary clinical evidence showing that the medicine may demonstrate substantial improvement compared to currently available therapies. This is known as Breakthrough Therapy Designation. Benefits are the same as PRIME.
5. Scientific advice
The EMA can provide advice to medicines developers on the most appropriate way to generate robust data on a medicine’s benefits and risks. A scientific advice can be requested at any stage of the development to ensure sponsors perform appropriate tests and studies so that no major objections are raised at the time of marketing authorization application.
When a sponsor is eligible to a scientific advice, a document gathering all questions the sponsor would like to ask has to be prepared and sent to the EMA within an agreed timeline. The questions can cover the quality aspects, non-clinical aspects, clinical aspects, and methodological issues. Responses or solutions to these questions must be included in the document. The document is then reviewed by the Scientific Advice Working Party (SAWP) of the EMA to give advice on the sponsor’s proposals and make a recommendation. The final advice is given by the Committee for Medicinal products for Human Use (CHMP) to the sponsor.
A scientific advice does not pre-evaluate results of studies and does not conclude on whether the benefits of the medicine outweigh the risks. A scientific advice is not legally binding.
Scientific advice meetings can be requested by any medicine developers. They are particularly helpful for academic and micro, small and medium sized companies who have a limited knowledge of medicine regulations.
Applicants pay administrative fee for a scientific advice. Micro, small and medium sized companies have a 90% fee reduction.
II. Early Access to the EU market

1. Centralised procedure
The use of the centralised procedure is mandatory to market an ATMP in the EU as laid down in Regulation (EC) No 726/2004. The centralized procedure is the Union-wide procedure with a single application, a single evaluation and a single authorization throughout the EU.
ATMP applications are reviewed and assessed by the CAT. Experts of the CAT assess quality, safety, and efficacy data to ensure benefits of the medicine outweigh the risks. The CAT opinion is then sent to the CHMP who will provide a recommendation on whether the ATMP should be authorized or not. The favorable opinion or unfavorable opinion is then sent to the European Commission to approve or reject the marketing authorization.
Several regulatory pathways can be followed to successfully market your ATMP in Europe. They are summarized in the below figure.
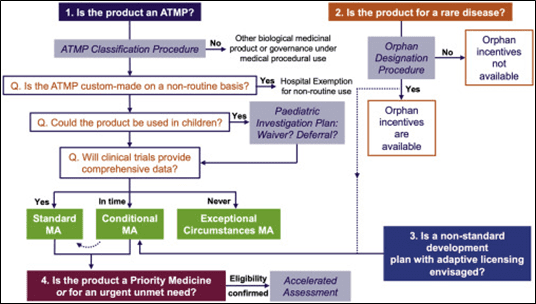
Figure 2: EU regulatory pathways for ATMPs3
2. PRIME scheme and accelerated assessment
Once the PRIME designation has been granted to a medicine, one of the benefits is to be eligible for accelerated assessment at the time of marketing authorization application. Applicants for accelerated assessment should justify the fact that the medicine is expected to be a major public health interest particularly from the point of view of therapeutic innovation.
Sponsors wishing to apply for an accelerated assessment should send a request to the EMA at least two or three months before submitting their MAA.
Accelerated assessment reduces the time of assessment by the CAT and CHMP from 210 days (standard MAA evaluation) to 150 days.
Once the Breakthrough Therapy designation has been granted to a medicine by the FDA, submission of portions of data before marketing authorization application is possible. This is the rolling review process.
The table below shows the mains differences between the PRIME designation and the Breakthrough Therapy designation.
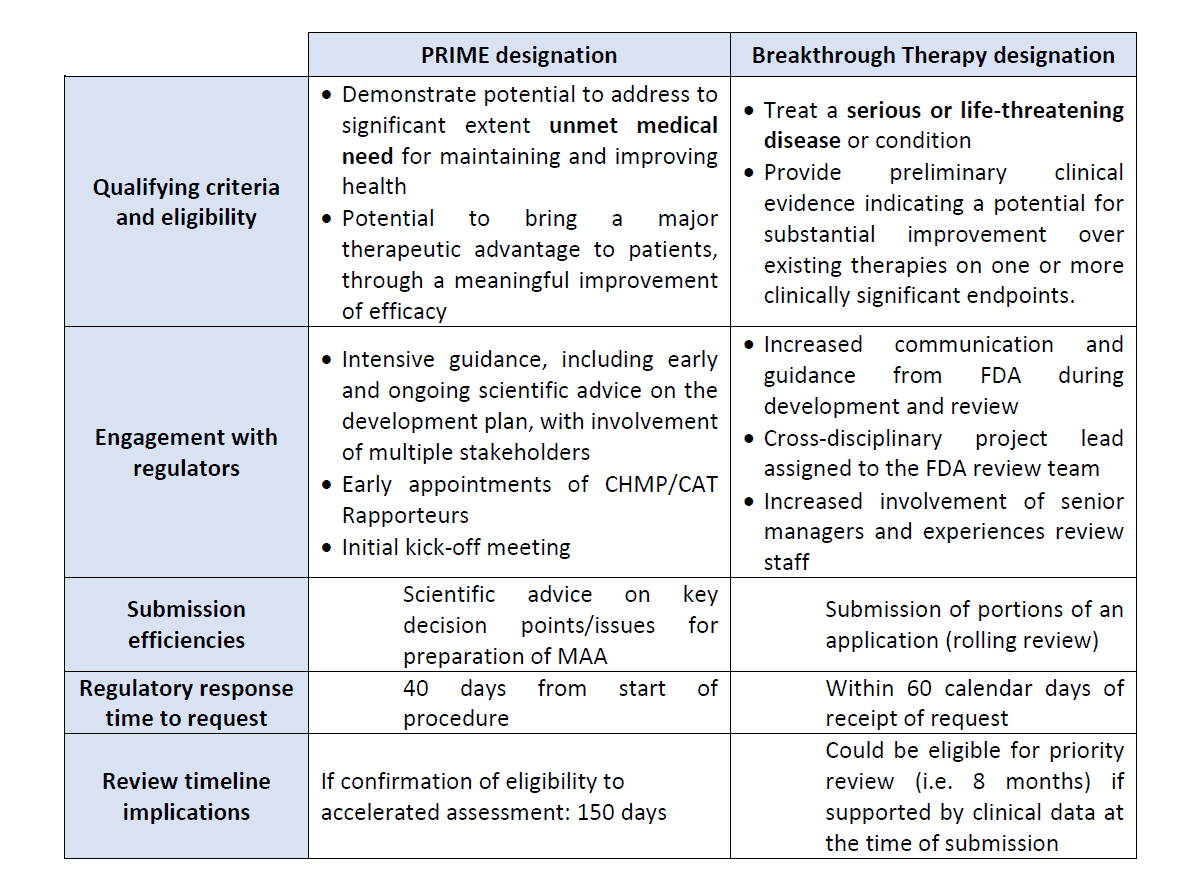
Figure 3: PRIME scheme and Breakthrough designation4
3. Conditional MA
A conditional MA can be granted by the EMA to ATMPs in the interest of public health. These products have less comprehensive clinical data than normally required but the benefits of immediate availability of the medicines outweighs the risk inherent in the fact that additional data are still required. The following criteria have to be met to be eligible to conditional marketing authorisation: the benefit/risks ratio of the medicine is positive, it is likely that the applicant will be able to provide comprehensive data post-authorization, the medicine fulfills an unmet medical need and the benefit of the medicine’s immediate availability to patients is greater than the risk inherent in the fact that additional data are required.
Once the MA is granted, the MAH must fulfills specific obligations within defined timelines.
The conditional marketing authorization is valid for one year and can be renewed annually.
The conditional marketing authorization can be converted into a standard marketing authorisation once all specific obligations are fulfilled and the complete data confirm that the benefits continue to outweigh the risks. The standard marketing authorization is valid for five years and can be renewed for unlimited validity.
III. How can BlueReg help?

At BlueReg, our team of experts has an excellent knowledge of the EU regulation and an excellent understanding of the scientific and development challenges that developers can face with innovative medicines including ATMPs. Our regulatory experts also have an excellent understanding of EMA expectations for these products and can help medicine developers defining the best suitable regulatory strategy.
We can advise developers on very early and late stage of the development process on several innovative medicines. We work with sponsors on the risk-based approach and we select the appropriate tools at the right time to design the best regulatory strategy and get a successful marketing authorization.
Blue Reg has a keen interest in innovative medicines and worked on many innovative medicines so far. Experience gained from these projects is applicable to the issues you can face when developing an ATMP. We can guide you at every regulatory step described above and beyond (discussion on the risk-based approach, pediatric investigation plan, orphan drug designation and post-approval activities). Learn more about our services for ATMPs
Consultancy companies are very sought by non-European companies who do not have European affiliates. If SME companies are not established in the EU/EEA, SME incentives can be accessed through BlueReg regulatory consultancy who has the SME status. BlueReg and their EMA approved SME clients have access to several benefits such as administrative, regulatory, and financial support.
List of acronyms:
ATMP: Advanced Therapy Medicinal Product
CAT: Committee for Advanced Therapies
CHMP: Committee for Medicinal Products for Human Use
EMA: European Medicines Agency
EU: European Union
ITF: Innovation Task Force
MAA: Marketing Authorization Application
SWAP: Scientific Advice Working Group
References:
1. Hanna E, Remuzat C, Auquier P, et al. Advanced therapy medicinal products: current and future perspectives. J Mark Access Health Policy 2016
2 Romaldas Maciulaitis, Lucia D'Apote et al. Clinical Development of Advanced Therapy Medicinal Products in Europe: Evidence That Regulators Must Be Proactive. Research Gate. March 2012 – Available at: Clinical_Development_of_Advanced_Therapy_Medicinal.pdf
3 GiuliaDetela, AnthonyLodge. EU Regulatory Pathways for ATMPs: Standard, Accelerated and Adaptive Pathways to Marketing Authorisation. Science Direct. June 2019. Available at: https://www.sciencedirect.com/science/article/pii/S2329050119300130#mmc2
4 Susan Forda. EMA’s PRIME can help speed delivery of new medicines to patients. March 2016. Available at: https://www.efpia.eu/news-events/the-efpia-view/efpia-news/160301-prime-for-delivery-of-new-medicines/#
Written by Marie-Sophie Caplain - Regulatory Affairs Associate
Did you like this article? Share on social networks:



